Valorisation scientifique




Liste des publications impliquant le personnel de la plateforme depuis 2018
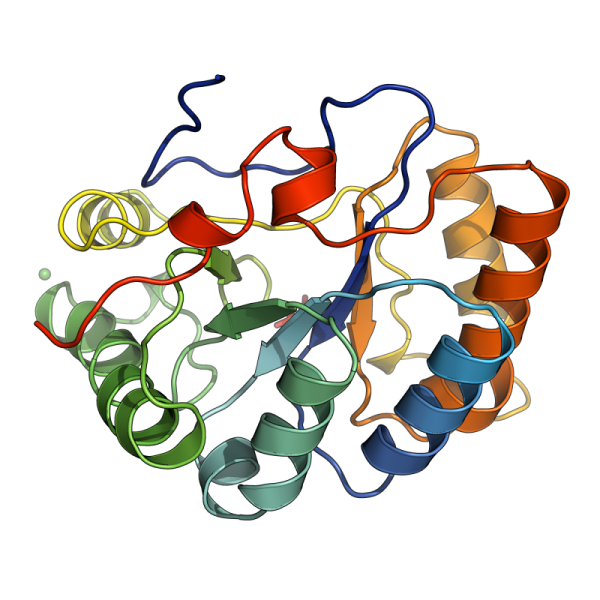
A single bacterial sulfatase is required for metabolism of colonic mucin O-glycans and intestinal colonization by a symbiotic human gut bacterium. Nature 2021 Oct;598(7880):332-337.
Humans have co-evolved with a dense community of microbial symbionts that inhabit the lower intestine. In the colon, secreted mucus creates a physical barrier that separates these microbes from the intestinal epithelium. Some gut bacteria are able to utilize mucin glycoproteins, the main mucus component, as a nutrient source. However, it remains unclear which bacterial enzymes initiate the degradation of the highly complex O-glycans found in mucins. In the colon, these glycans are heavily sulfated, but the specific sulfatases that are active on colonic mucins have not been identified. Here, we show that sulfatases are essential to the utilization of colonic mucin O-glycans by the human gut symbiont Bacteroides thetaiotaomicron. We have characterized the activity of 12 different sulfatases encoded by this species, showing that these enzymes collectively are active on all of the known sulfate linkages in colonic O-glycans. Crystal structures of 3 enzymes provide mechanistic insight into the molecular basis of substrate-specificity. Unexpectedly, we found that a single sulfatase is essential for utilization of sulfated O-glycans in vitro and also plays a major role in vivo. Our results provide insight into the mechanisms of mucin degradation by gut bacteria, an important process for both normal microbial gut colonization and diseases such as inflammatory bowel disease (IBD). Sulfatase activity is likely to be a keystone step in bacterial mucin degradation and inhibition of these enzymes may therefore represent a viable therapeutic path for treatment of IBD and other diseases.
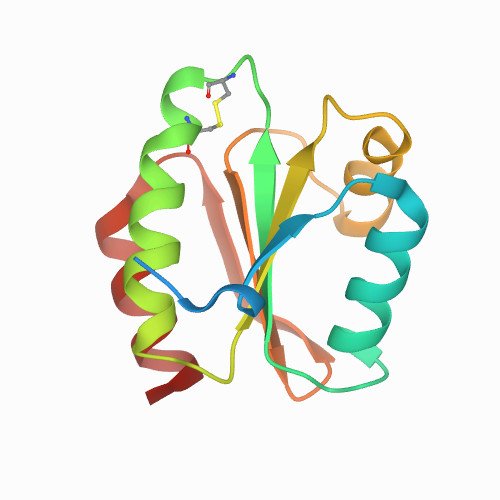
A novel thermostable prokaryotic fucoidan active sulfatase PsFucS1 with an unusual quaternary hexameric structure. Scientific Reports 2021 Sep 30;11(1):19523.
Fucoidans are sulfated, fucose-rich marine polysaccharides primarily found in cell walls of brown seaweeds (macroalgae). Fucoidans are known to possess beneficial bioactivities depending on their structure and sulfation degree. Here, we report the first functional characterization and the first crystal structure of a prokaryotic sulfatase, PsFucS1, belonging to sulfatase subfamily S1_13, able to release sulfate from fucoidan oligosaccharides. PsFucS1 was identified in the genome of a Pseudoalteromonas sp. isolated from sea cucumber gut. PsFucS1 (57 kDa) is Ca2+ dependent and has an unusually high optimal temperature (68 °C) and thermostability. Further, the PsFucS1 displays a unique quaternary hexameric structure comprising a tight trimeric dimer complex. The structural data imply that this hexamer formation results from an uncommon interaction of each PsFucS1 monomer that is oriented perpendicular to the common dimer interface (~ 1500 Å2) that can be found in analogous sulfatases. The uncommon interaction involves interfacing (1246 Å2) through a bundle of α-helices in the N-terminal domain to form a trimeric ring structure. The high thermostability may be related to this unusual quaternary hexameric structure formation that is suggested to represent a novel protein thermostabilization mechanism.
In silico and in vitro analysis of an Aspergillus niger chitin deacetylase to decipher its subsite sugar preferences. Journal of Biological Chemistry 2021 Sep 1;297(4):101129.
Chitin deacetylases (CDAs) are found in many different organisms ranging from marine bacteria to fungi and insects. These enzymes catalyze the removal of acetyl groups from chitinous substrates generating various chitosans, linear co- polymers consisting of N-acetylglucosamine (GlcNAc) and glucosamine (GlcN). CDAs influence the degree of acetylation (DA) of chitosans as well as their pattern of acetylation (PA), a parameter which was recently shown to influence the physicochemical properties and biological activities of chitosans. The binding site of CDAs typically consists of around four subsites, each accommodating a single sugar unit of the substrate. It has been hypothesized that the subsite preferences for GlcNAc or GlcN units play a crucial role in the acetylation pattern they generate, but so far, this characteristic was largely ignored, and still lacks structural data on the involved residues.
Here, we determined the crystal structure of an Aspergillus niger CDA (AngCDA). Then, we used molecular dynamics simulations, backed up with a variety of in vitro activity assays using different well- defined polymeric and oligomeric substrates, to study this CDA in detail. We found that AngCDA strongly prefers a GlcNAc sugar unit at its -1 subsite and shows a weak GlcNAc preference at the other non-catalytic subsites, which was apparent both when de- and N- acetylating oligomeric substrates. Overall, our results show that the combination of in vitro and in silico methods used here enables the detailed analysis of CDAs, including their subsite preferences, which could influence their substrate targets and the characteristics of chitosans produced by these species.
Structure–function analysis of a new PL17 oligoalginate lyase from the marine bacterium Zobellia galactanivorans DsijT. Glycobiology 2021 Jun 28;cwab058.
Alginate is a major compound of brown macroalgae and as such an important carbon and energy source for heterotrophic marine bacteria. Despite the rather simple composition of alginate only comprising mannuronate and guluronate units, these bacteria feature complex alginolytic systems that can contain up to seven alginate lyases. This reflects the necessity of large enzyme systems for the complete degradation of the abundant substrate. Numerous alginate lyases have been characterized. They belong to different polysaccharide lyase (PL) families, but only one crystal structure of a family 17 (PL17) alginate lyase has been reported to date, namely Alg17c from the gammaproteobacterium Saccharophagus degradans. Biochemical and structural characterizations are helpful to link sequence profiles to function, evolution of functions and nichespecific characteristics. Here, we combined detailed biochemical and crystallographic analysis of AlyA3, a PL17 alginate lyase from the marine flavobacteria Zobellia galactanivorans DsijT, providing the first structure of a PL17 in the Bacteroidetes phylum. AlyA3 is exo-lytic and highly specific of mannuronate stretches. As part of an “alginate utilizing locus”, its activity is complementary to that of other characterized alginate lyases from the same bacterium. Structural comparison with Alg17c highlights a common mode of action for exo-lytic cleavage of the substrate, strengthening our understanding of the PL17 catalytic mechanism. We show that unlike Alg17c, AlyA3 contains an inserted flexible loop at the entrance to the catalytic groove, likely involved in substrate recognition, processivity and turn over.
Atypical Iron-Sulfur Cluster Binding, Redox Activity and Structural Properties of Chlamydomonas reinhardtii Glutaredoxin 2. Antioxidants (Basel) 2021 May 19;10(5):803.
Glutaredoxins (GRXs) are thioredoxin superfamily members exhibiting thiol-disulfide oxidoreductase activity and/or iron-sulfur (Fe-S) cluster binding capacities. These properties are determined by specific structural factors. In this study, we examined the capacity of the class I Chlamydomonas reinhardtii GRX2 recombinant protein to catalyze both protein glutathionylation and deglutathionylation reactions using a redox sensitive fluorescent protein as a model protein substrate. We observed that the catalytic cysteine of the CPYC active site motif of GRX2 was sufficient for catalyzing both reactions in the presence of glutathione. Unexpectedly, spectroscopic characterization of the protein purified under anaerobiosis showed the presence of a [2Fe-2S] cluster despite having a presumably inadequate active site signature, based on past mutational analyses. The spectroscopic characterization of cysteine mutated variants together with modeling of the Fe–S cluster-bound GRX homodimer from the structure of an apo-GRX2 indicate the existence of an atypical Fe–S cluster environment and ligation mode. Overall, the results further delineate the biochemical and structural properties of conventional GRXs, pointing to the existence of multiple factors more complex than anticipated, sustaining the capacity of these proteins to bind Fe–S clusters.

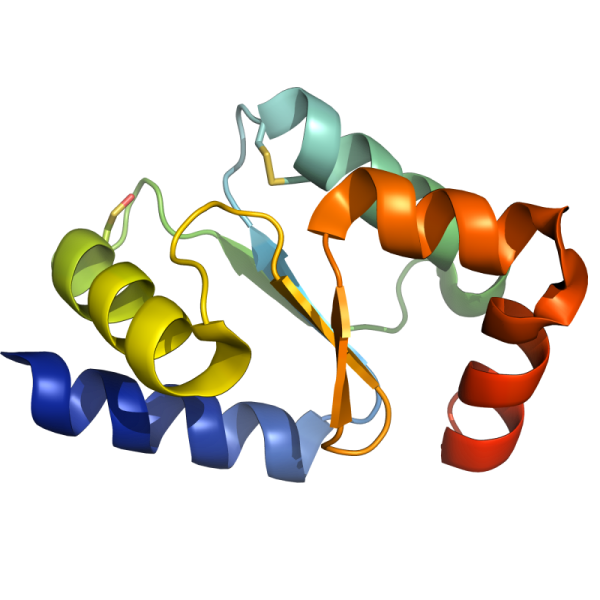
Sinorhizobium meliloti YrbA binds divalent metal cations using two conserved histidines. Bioscience Reports 2020 Oct 30;40(10):BSR20202956.
Sinorhizobium meliloti is a nitrogen-fixing bacterium forming symbiotic nodules with the legume Medicago truncatula. S. meliloti possesses two BolA-like proteins (BolA and YrbA), the function of which is unknown. In organisms where BolA proteins and monothiol glutaredoxins (Grxs) are present, they contribute to the regulation of iron homeostasis by bridging a [2Fe-2S] cluster into heterodimers. A role in the maturation of iron-sulfur (Fe-S) proteins is also attributed to both proteins. In the present study, we have performed a structure-function analysis of SmYrbA showing that it coordinates diverse divalent metal ions (Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) using His32 and His67 residues, that are also used for Fe-S cluster binding in BolA-Grx heterodimers. It also possesses the capacity to form heterodimers with the sole monothiol glutaredoxin (SmGrx2) present in this species. Using cellular approaches analyzing the metal tolerance of S. meliloti mutant strains inactivated in the yrbA and/or bolA genes, we provide evidence for a connection of YrbA with the regulation of iron homeostasis. The mild defects in M. truncatula nodulation reported for the yrbA bolA mutant as compared with the stronger defects in nodule development previously observed for a grx2 mutant suggest functions independent of SmGrx2. These results help in clarifying the physiological role of BolA-type proteins in bacteria.

Structural and enzymatic characterisation of the Type III effector NopAA (=GunA) from Sinorhizobium fredii USDA257 reveals a Xyloglucan hydrolase activity. Scientific Reports 2020 Jun 18;10(1):9932.
Rhizobia are nitrogen-fixing soil bacteria that can infect legume plants to establish root nodules symbiosis. To do that, a complex exchange of molecular signals occurs between plants and bacteria. Among them, rhizobial Nops (Nodulation outer proteins), secreted by a type III secretion system (T3SS) determine the host-specificity for efficient symbiosis with plant roots. Little is known about the molecular function of secreted Nops (also called effectors (T3E)) and their role in the symbiosis process. We performed the structure-function characterization of NopAA, a T3E from Sinorhizobium fredii by using a combination of X-ray crystallography, biochemical and biophysical approaches. This work displays for the first time a complete structural and biochemical characterization of a symbiotic T3E. Our results showed that NopAA has a catalytic domain with xyloglucanase activity extended by a N-terminal unfolded secretion domain that allows its secretion. We proposed that these original structural properties combined with the specificity of NopAA toward xyloglucan, a key component of root cell wall which is also secreted by roots in the soil, can give NopAA a strategic position to participate in recognition between bacteria and plant roots and to intervene in nodulation process.
Target Of Rapamycin pathway in the white-rot fungus Phanerochaete chrysosporium. PLoS One 2020 Feb 20;15(2):e0224776.
The Target Of Rapamycin (TOR) signaling pathway is known to regulate growth in response to nutrient availability and stress in eukaryotic cells. In the present study, we have investigated the TOR pathway in the white-rot fungus Phanerochaete chrysosporium. Inhibition of TOR activity by rapamycin affects conidia germination and hyphal growth highlighting the conserved mechanism of susceptibility to rapamycin. Interestingly, the secreted protein content is also affected by the rapamycin treatment. Finally, homologs of the components of TOR pathway can be identified in P. chrysosporium. Altogether, those results indicate that the TOR pathway of P. chrysosporium plays a central role in this fungus.
A subfamily roadmap of the evolutionarily diverse glycoside hydrolase family 16 (GH16). Journal of Biological Chemistry 2019 Nov 1;294(44):15973-15986.
Glycoside hydrolase family (GH) 16 comprises a large and taxonomically diverse family of glycosidases and transglycosidases that adopt a common β-jelly-roll fold and are active on a range of terrestrial and marine polysaccharides. Presently, broadly insightful sequence-function correlations in GH16 are hindered by a lack of a systematic subfamily structure. To fill this gap, we have used a highly scalable protein sequence similarity network analysis to delineate nearly 23,000 GH16 sequences into 23 robust subfamilies, which are strongly supported by hidden Markov model and maximum likelihood molecular phylogenetic analyses. Subsequent evaluation of over 40 experimental three-dimensional structures has highlighted key tertiary structural differences, predominantly manifested in active-site loops, that dictate substrate specificity across the GH16 evolutionary landscape. As for other large GH families (i.e. GH5, GH13, and GH43), this new subfamily classification provides a roadmap for functional glycogenomics that will guide future bioinformatics and experimental structure-function analyses. The GH16 subfamily classification is publicly available in the CAZy database. The sequence similarity network workflow used here, SSNpipe, is freely available from GitHub.
A marine bacterial enzymatic cascade degrades the algal polysaccharide ulvan. Nature Chemical Biology 2019 Aug;15(8):803-812.
Marine seaweeds increasingly grow into extensive algal blooms, which are detrimental to coastal ecosystems, tourism and aquaculture. However, algal biomass is also emerging as a sustainable raw material for the bioeconomy. The potential exploitation of algae is hindered by our limited knowledge of the microbial pathways-and hence the distinct biochemical functions of the enzymes involved-that convert algal polysaccharides into oligo- and monosaccharides. Understanding these processes would be essential, however, for applications such as the fermentation of algal biomass into bioethanol or other value-added compounds. Here, we describe the metabolic pathway that enables the marine flavobacterium Formosa agariphila to degrade ulvan, the main cell wall polysaccharide of bloom-forming Ulva species. The pathway involves 12 biochemically characterized carbohydrate-active enzymes, including two polysaccharide lyases, three sulfatases and seven glycoside hydrolases that sequentially break down ulvan into fermentable monosaccharides. This way, the enzymes turn a previously unexploited renewable into a valuable and ecologically sustainable bioresource.
The agar-specific hydrolase Zg AgaC from the marine bacterium Zobellia galactanivorans defines a new GH16 protein subfamily. Journal of Biological Chemistry 2019 Apr 26;294(17):6923-6939.
Agars are sulfated galactans from red macroalgae and are composed of a d-galactose (G unit) and l-galactose (L unit) alternatively linked by α-1,3 and β-1,4 glycosidic bonds. These polysaccharides display high complexity, with numerous modifications of their backbone (e.g. presence of a 3,6-anhydro-bridge (LA unit) and sulfations and methylation). Currently, bacterial polysaccharidases that hydrolyze agars (β-agarases and β-porphyranases) have been characterized on simple agarose and more rarely on porphyran, a polymer containing both agarobiose (G-LA) and porphyranobiose (GL6S) motifs. How bacteria can degrade complex agars remains therefore an open question. Here, we studied an enzyme from the marine bacterium Zobellia galactanivorans (ZgAgaC) that is distantly related to the glycoside hydrolase 16 (GH16) family β-agarases and β-porphyranases. Using a large red algae collection, we demonstrate that ZgAgaC hydrolyzes not only agarose but also complex agars from Ceramiales species. Using tandem MS analysis, we elucidated the structure of a purified hexasaccharide product, L6S-G-LA2Me-G(2Pentose)-LA2S-G, released by the activity of ZgAgaC on agar extracted from Osmundea pinnatifida By resolving the crystal structure of ZgAgaC at high resolution (1.3 Å) and comparison with the structures of ZgAgaB and ZgPorA in complex with their respective substrates, we determined that ZgAgaC recognizes agarose via a mechanism different from that of classical β-agarases. Moreover, we identified conserved residues involved in the binding of complex oligoagars and demonstrate a probable influence of the acidic polysaccharide's pH microenvironment on hydrolase activity. Finally, a phylogenetic analysis supported the notion that ZgAgaC homologs define a new GH16 subfamily distinct from β-porphyranases and classical β-agarases.
Asp271 is critical for substrate interaction with the surface binding site in β-agarase a from Zobellia galactanivorans. Proteins 2019 Jan;87(1):34-40.
In the marine environment agar degradation is assured by bacteria that contain large agarolytic systems with enzymes acting in various endo- and exo-modes. Agarase A (AgaA) is an endo-glycoside hydrolase of family 16 considered to initiate degradation of agarose. Agaro-oligosaccharide binding at a unique surface binding site (SBS) in AgaA from Zobellia galactanivorans was investigated by computational methods in conjunction with a structure/sequence guided approach of site-directed mutagenesis probed by surface plasmon resonance binding analysis of agaro-oligosaccharides of DP 4-10. The crystal structure has shown that agaro-octaose interacts via H-bonds and aromatic stacking along 7 subsites (L through R) of the SBS in the inactive catalytic nucleophile mutant AgaA-E147S. D271 is centrally located in the extended SBS where it forms H-bonds to galactose and 3,6-anhydrogalactose residues of agaro-octaose at subsites O and P. We propose D271 is a key residue in ligand binding to the SBS. Thus AgaA-E147S/D271A gave slightly decreasing KD values from 625 ± 118 to 468 ± 13 μM for agaro-hexaose, -octaose, and -decaose, which represent 3- to 4-fold reduced affinity compared with AgaA-E147S. Molecular dynamics simulations and interaction analyses of AgaA-E147S/D271A indicated disruption of an extended H-bond network supporting that D271 is critical for the functional SBS. Notably, neither AgaA-E147S/W87A nor AgaA-E147S/W277A, designed to eliminate stacking with galactose residues at subsites O and Q, respectively, were produced in soluble form. W87 and W277 may thus control correct folding and structural integrity of AgaA.
The laterally acquired GH5 Zg EngA GH5_4 from the marine bacterium Zobellia galactanivorans is dedicated to hemicellulose hydrolysis. Biochemal Journal 2018 Nov 28;475(22):3609-3628.
Cell walls of marine macroalgae are composed of diverse polysaccharides that provide abundant carbon sources for marine heterotrophic bacteria. Among them, Zobellia galactanivorans is considered as a model for studying algae-bacteria interactions. The degradation of typical algal polysaccharides, such as agars or alginate, has been intensively studied in this model bacterium, but the catabolism of plant-like polysaccharides is essentially uncharacterized. Here, we identify a polysaccharide utilization locus in the genome of Z. galactanivorans, induced by laminarin (β-1,3-glucans), and containing a putative GH5 subfamily 4 (GH5_4) enzyme, currently annotated as a endoglucanase (ZgEngAGH5_4). A phylogenetic analysis indicates that ZgEngAGH5_4 was laterally acquired from an ancestral Actinobacteria We performed the biochemical and structural characterization of ZgEngAGH5_4 and demonstrated that this GH5 is, in fact, an endo-β-glucanase, most active on mixed-linked glucan (MLG). Although ZgEngAGH5_4 and GH16 lichenases both hydrolyze MLG, these two types of enzymes release different series of oligosaccharides. Structural analyses of ZgEngAGH5_4 reveal that all the amino acid residues involved in the catalytic triad and in the negative glucose-binding subsites are conserved, when compared with the closest relative, the cellulase EngD from Clostridium cellulovorans, and some other GH5s. In contrast, the positive glucose-binding subsites of ZgEngAGH5_4 are different and this could explain the preference for MLG, with respect to cellulose or laminarin. Molecular dynamics computer simulations using different hexaoses reveal that the specificity for MLG occurs through the +1 and +2 subsites of the binding pocket that display the most important differences when compared with the structures of other GH5_4 enzymes.
Discovery and screening of novel metagenome-derived GH107 enzymes targeting sulfated fucans from brown algae. FEBS Journal 2018 Nov;285(22):4281-4295.
Sulfated fucans, often denoted as fucoidans, are highly variable cell wall polysaccharides of brown algae, which possess a wide range of bioactive properties with potential pharmaceutical applications. Due to their complex architecture, the structures of algal fucans have until now only been partly determined. Enzymes capable of hydrolyzing sulfated fucans may allow specific release of defined bioactive oligosaccharides and may serve as a tool for structural elucidation of algal walls. Currently, such enzymes include only a few hydrolases belonging to the glycoside hydrolase family 107 (GH107), and little is known about their mechanistics and the substrates they degrade. In this study, we report the identification and recombinant production of three novel GH107 family proteins derived from a marine metagenome. Activity screening against a large substrate collection showed that all three enzymes degraded sulfated fucans from brown algae in the order Fucales. This is in accordance with a hydrolytic activity against α-1,4-fucosidic linkages in sulfated fucans as reported for previous GH107 members. Also, the activity screening gave new indications about the structural differences in brown algal cell walls. Finally, sequence analyses allowed identification of the proposed catalytic residues of the GH107 family. The findings presented here form a new basis for understanding the GH107 family of enzymes and investigating the complex sulfated fucans from brown algae. DATABASE: The assembled metagenome and raw sequence data is available at EMBL-EBI (Study number: PRJEB28480). Sequences of the GH107 fucanases (Fp273, Fp277, and Fp279) have been deposited in GenBank under accessions MH755451-MH755453.

Mitochondrial Arabidopsis thaliana TRXo Isoforms Bind an Iron⁻Sulfur Cluster and Reduce NFU Proteins In Vitro. Antioxidants (Basel) 2018 Oct 13;7(10):142.
In plants, the mitochondrial thioredoxin (TRX) system generally comprises only one or two isoforms belonging to the TRX h or o classes, being less well developed compared to the numerous isoforms found in chloroplasts. Unlike most other plant species, Arabidopsis thaliana possesses two TRXo isoforms whose physiological functions remain unclear. Here, we performed a structure⁻function analysis to unravel the respective properties of the duplicated TRXo1 and TRXo2 isoforms. Surprisingly, when expressed in Escherichia coli, both recombinant proteins existed in an apo-monomeric form and in a homodimeric iron⁻sulfur (Fe-S) cluster-bridged form. In TRXo2, the [4Fe-4S] cluster is likely ligated in by the usual catalytic cysteines present in the conserved Trp-Cys-Gly-Pro-Cys signature. Solving the three-dimensional structure of both TRXo apo-forms pointed to marked differences in the surface charge distribution, notably in some area usually participating to protein⁻protein interactions with partners. However, we could not detect a difference in their capacity to reduce nitrogen-fixation-subunit-U (NFU)-like proteins, NFU4 or NFU5, two proteins participating in the maturation of certain mitochondrial Fe-S proteins and previously isolated as putative TRXo1 partners. Altogether, these results suggest that a novel regulation mechanism may prevail for mitochondrial TRXs o, possibly existing as a redox-inactive Fe-S cluster-bound form that could be rapidly converted in a redox-active form upon cluster degradation in specific physiological conditions.
Comparison of photosynthetic performances of marine picocyanobacteria with different configurations of the oxygen-evolving complex. Photosynthesis Research 2018 Oct;138(1):57-71.
The extrinsic PsbU and PsbV proteins are known to play a critical role in stabilizing the Mn4CaO5 cluster of the PSII oxygen-evolving complex (OEC). However, most isolates of the marine cyanobacterium Prochlorococcus naturally miss these proteins, even though they have kept the main OEC protein, PsbO. A structural homology model of the PSII of such a natural deletion mutant strain (P. marinus MED4) did not reveal any obvious compensation mechanism for this lack. To assess the physiological consequences of this unusual OEC, we compared oxygen evolution between Prochlorococcus strains missing psbU and psbV (PCC 9511 and SS120) and two marine strains possessing these genes (Prochlorococcus sp. MIT9313 and Synechococcus sp. WH7803). While the low light-adapted strain SS120 exhibited the lowest maximal O2 evolution rates (Pmax per divinyl-chlorophyll a, per cell or per photosystem II) of all four strains, the high light-adapted strain PCC 9511 displayed even higher PChlmax and PPSIImax at high irradiance than Synechococcus sp. WH7803. Furthermore, thermoluminescence glow curves did not show any alteration in the B-band shape or peak position that could be related to the lack of these extrinsic proteins. This suggests an efficient functional adaptation of the OEC in these natural deletion mutants, in which PsbO alone is seemingly sufficient to ensure proper oxygen evolution. Our study also showed that Prochlorococcus strains exhibit negative net O2 evolution rates at the low irradiances encountered in minimum oxygen zones, possibly explaining the very low O2 concentrations measured in these environments, where Prochlorococcus is the dominant oxyphototroph.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
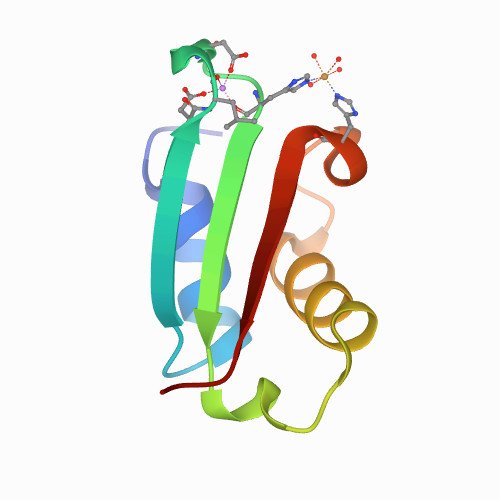
Trametes versicolor glutathione transferase Xi 3, a dual Cys‐GST with catalytic specificities of both Xi and Omega classes. FEBS Letters 2018 Sep;592(18):3163-3172.
Glutathione transferases (GSTs) from the Xi and Omega classes have a catalytic cysteine residue, which gives them reductase activities. Until now, they have been assigned distinct substrates. While Xi GSTs specifically reduce glutathionyl-(hydro)quinones, Omega GSTs are specialized in the reduction of glutathionyl-acetophenones. Here, we present the biochemical and structural analysis of TvGSTX1 and TvGSTX3 isoforms from the wood-degrading fungus Trametes versicolor. TvGSTX1 reduces GS-menadione as expected, while TvGSTX3 reduces both Xi and Omega substrates. An in-depth structural analysis indicates a broader active site for TvGSTX3 due to specific differences in the nature of the residues situated in the C-terminal helix α9. This feature could explain the catalytic duality of TvGSTX3. Based on phylogenetic analysis, we propose that this duality might exist in saprophytic fungi and ascomycetes.
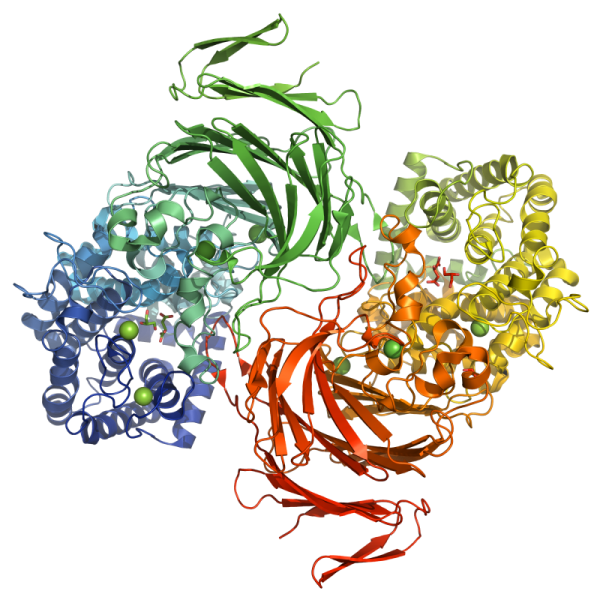
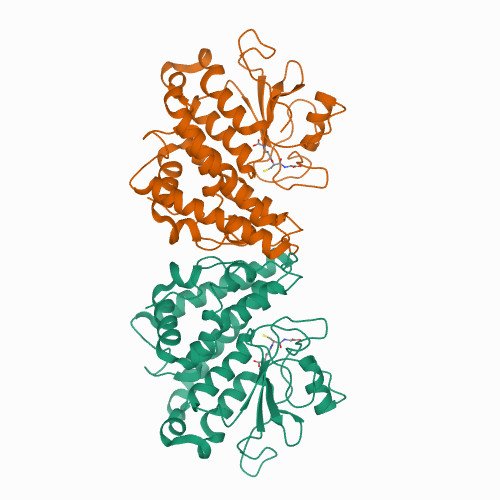
X-ray Diffraction and Density Functional Theory Provide Insight into Vanadate Binding to Homohexameric Bromoperoxidase II and the Mechanism of Bromide Oxidation. ACS Chemical Biology 2018 May 18;13(5):1243-1259.
X-ray diffraction of native bromoperoxidase II (EC 1.11.1.18) from the brown alga Ascophyllum nodosum reveals at a resolution of 2.26 Å details of orthovanadate binding and homohexameric protein organization. Three dimers interwoven in contact regions and tightened by hydrogen-bond-clamped guanidinium stacks along with regularly aligned water molecules form the basic structure of the enyzme. Intra- and intermolecular disulfide bridges further stabilize the enzyme preventing altogether the protein from denaturing up to a temperature of 90 °C, as evident from dynamic light scattering and the on-gel ortho-dianisidine assay. Every monomer binds one equivalent of orthovanadate in a cavity formed from side chains of three histidines, two arginines, one lysine, serine, and tryptophan. Protein binding occurs primarily through hydrogen bridges and superimposed by Coulomb attraction according to thermochemical model on density functional level of theory (B3LYP/6-311++G**). The strongest attractor is the arginine side chain mimic N-methylguanidinium, enhancing in positive cooperative manner hydrogen bridges toward weaker acceptors, such as residues from lysine and serine. Activating hydrogen peroxide occurs in the thermochemical model by side-on binding in orthovanadium peroxoic acid, oxidizing bromide with virtually no activation energy to hydrogen bonded hypobromous acid.
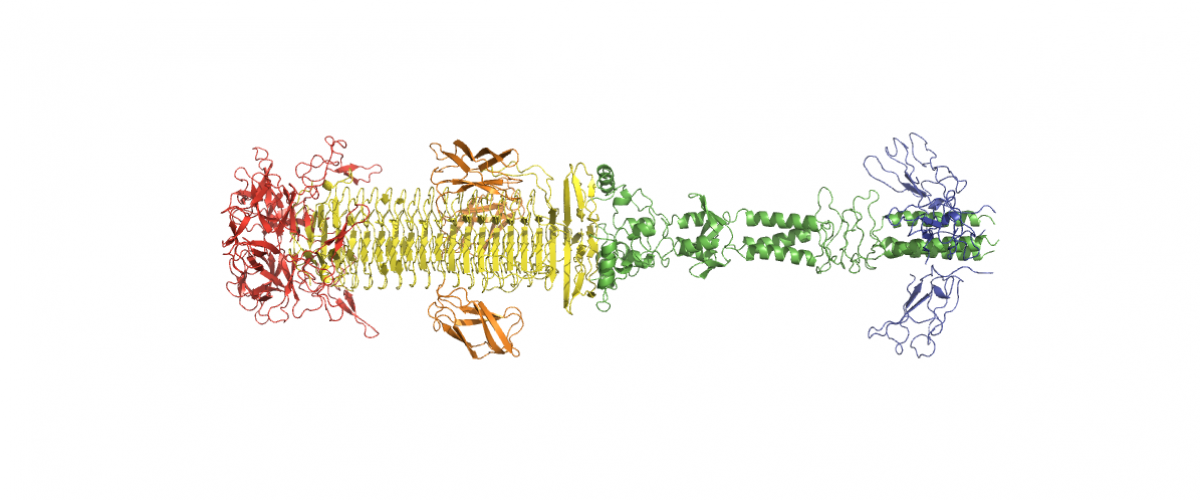
Dystrophin's central domain forms a complex filament that becomes disorganized by in-frame deletions. Journal of Biological Chemistry 2018 May 4;293(18):6637-6646.
Dystrophin, encoded by the DMD gene, is critical for maintaining plasma membrane integrity during muscle contraction events. Mutations in the DMD gene disrupting the reading frame prevent dystrophin production and result in severe Duchenne muscular dystrophy (DMD); in-frame internal deletions allow production of partly functional internally deleted dystrophin and result in less severe Becker muscular dystrophy (BMD). Many known BMD deletions occur in dystrophin's central domain, generally considered to be a monotonous rod-shaped domain based on the knowledge of spectrin family proteins. However, the effects caused by these deletions, ranging from asymptomatic to severe BMD, argue against the central domain serving only as a featureless scaffold. We undertook structural studies combining small-angle X-ray scattering and molecular modeling in an effort to uncover the structure of the central domain, as dystrophin has been refractory to characterization. We show that this domain appears to be a tortuous and complex filament that is profoundly disorganized by the most severe BMD deletion (loss of exons 45-47). Despite the preservation of large parts of the binding site for neuronal nitric oxide synthase (nNOS) in this deletion, computational approaches failed to recreate the association of dystrophin with nNOS. This observation is in agreement with a strong decrease of nNOS immunolocalization in muscle biopsies, a parameter related to the severity of BMD phenotypes. The structural description of the whole dystrophin central domain we present here is a first necessary step to improve the design of microdystrophin constructs toward the goal of a successful gene therapy for DMD.
A Novel Enzyme Portfolio for Red Algal Polysaccharide Degradation in the Marine Bacterium Paraglaciecola hydrolytica S66 T Encoded in a Sizeable Polysaccharide Utilization Locus. Frontiers in Microbiology 2018 May 3;9:839.
Marine microbes are a rich source of enzymes for the degradation of diverse polysaccharides. Paraglaciecola hydrolytica S66T is a marine bacterium capable of hydrolyzing polysaccharides found in the cell wall of red macroalgae. In this study, we applied an approach combining genomic mining with functional analysis to uncover the potential of this bacterium to produce enzymes for the hydrolysis of complex marine polysaccharides. A special feature of P. hydrolytica S66T is the presence of a large genomic region harboring an array of carbohydrate-active enzymes (CAZymes) notably agarases and carrageenases. Based on a first functional characterization combined with a comparative sequence analysis, we confirmed the enzymatic activities of several enzymes required for red algal polysaccharide degradation by the bacterium. In particular, we report for the first time, the discovery of novel enzyme activities targeting furcellaran, a hybrid carrageenan containing both β-carrageenan and κ/β-carrageenan motifs. Some of these enzymes represent a new subfamily within the CAZy classification. From the combined analyses, we propose models for the complete degradation of agar and κ/β-type carrageenan by P. hydrolytica S66T. The novel enzymes described here may find value in new bio-based industries and advance our understanding of the mechanisms responsible for recycling of red algal polysaccharides in marine ecosystems.
Double blind microarray-based polysaccharide profiling enables parallel identification of uncharacterized polysaccharides and carbohydrate-binding proteins with unknown specificities. Scientific Reports 2018 Feb 6;8(1):2500.
Marine algae are one of the largest sources of carbon on the planet. The microbial degradation of algal polysaccharides to their constitutive sugars is a cornerstone in the global carbon cycle in oceans. Marine polysaccharides are highly complex and heterogeneous, and poorly understood. This is also true for marine microbial proteins that specifically degrade these substrates and when characterized, they are frequently ascribed to new protein families. Marine (meta)genomic datasets contain large numbers of genes with functions putatively assigned to carbohydrate processing, but for which empirical biochemical activity is lacking. There is a paucity of knowledge on both sides of this protein/carbohydrate relationship. Addressing this 'double blind' problem requires high throughput strategies that allow large scale screening of protein activities, and polysaccharide occurrence. Glycan microarrays, in particular the Comprehensive Microarray Polymer Profiling (CoMPP) method, are powerful in screening large collections of glycans and we described the integration of this technology to a medium throughput protein expression system focused on marine genes. This methodology (Double Blind CoMPP or DB-CoMPP) enables us to characterize novel polysaccharide-binding proteins and to relate their ligands to algal clades. This data further indicate the potential of the DB-CoMPP technique to accommodate samples of all biological sources.