[Publication] A method for comparing the metabolism of photosynthetic eukaryotes




Genome-scale metabolic networks make it possible to model all the known chemical transformations in the metabolism of a given organism. Comparing them on a large scale means overcoming the problems of heterogeneity among the available genomes. In an article published in Genome Research, scientists propose an approach for constructing networks of homogeneous quality that can be compared from an evolutionary perspective.
Over the last ten years or so, several maps of algal metabolism have been reconstructed from genome sequencing data. When the first attempts were made to compare those metabolic networks, the studies explained the differences observed between the metabolic maps by the heterogeneity of the sequencing data sets and not by any real biological differences between the species. The new methodological approach, implemented in the AuCoMe software (distributed under an open licence), was developed using comparative genomics. By building on the expert knowledge contained in the genomic annotations of the best described species, it is possible to deduce the metabolic maps of species whose genome annotations are more rudimentary - or incomplete. Missing genes have also been identified by searching for sequence similarities between related species. Finally, the method also incorporates an approach to guarantee the reliability of predictions by limiting the biases potentially induced by the specific features of a given genome. This method combines knowledge engineering techniques, genomic sequence comparisons and biological system modelling to simultaneously analyse and compare the metabolism of several eukaryotic species, whereas until now the state of the art has only been able to compare a few species.
This methodology has been applied to study a family of some forty very diverse algal genomes: several lineages of microalgae, including diatoms and cryptophytes, as well as green, red and brown macroalgae. It has produced a unique catalogue of metabolic maps for dozens of marine eukaryotes. Comparisons between these maps have shown that the differences between the metabolisms of the different species studied are essentially in line with the reference phylogeny, i.e. with the current state of knowledge about the relationships between these organisms. There are, however, some notable differences. In particular, the authors' predictions suggest that the microalga Guillardia theta, considered to belong to the cryptophyte group, whose phylogenetic position is still debated, has a metabolism that seems to share characteristics with both the archaeplastids (grouping green lineage and red algae) and stramenopiles (brown algae and diatoms).
This method provides a rapid initial overview of the metabolism of a given group of organisms, paving the way for more in-depth studies of certain specificities that require further experimental investigation.
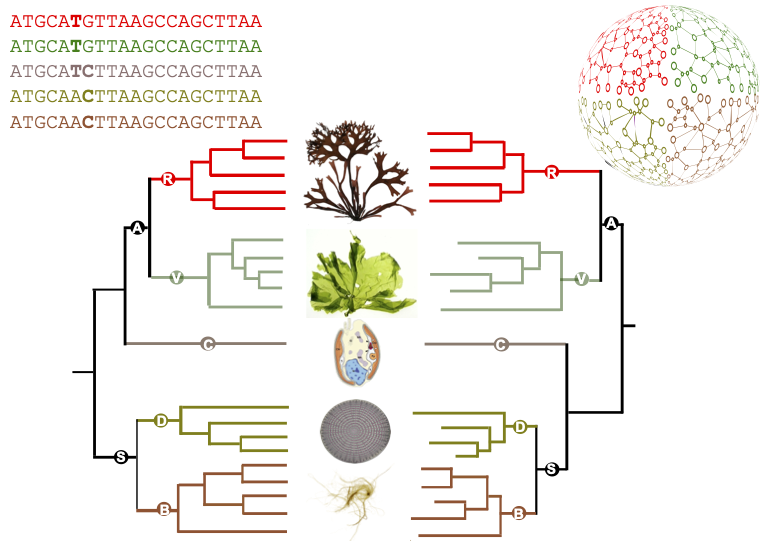
Figure : comparison between a reference phylogeny generated from sequences (left-hand side) and the corresponding metabolic dendrogram, resulting from the comparison of metabolic networks (right-hand side). Simplified from the article.
Figure credits: Ryo Onuma, Neha Mishra, Shin-ya Miyagishima, George Swann (CC-BY SA 4.0), Jeanne Got (Irisa), Jonas Collén, Simon Dittami, Gabriel Markov (Station Biologique Roscoff).
Crédits vignette : George Swann (CC-BY SA 4.0), Jeanne Got (Irisa), Jonas Collén, Simon Dittami, Gabriel Markov (Station Biologique Roscoff).
To find out more, please read:
Inferring and comparing metabolism across heterogeneous sets of annotated genomes using AuCoMe
Arnaud Belcour, Jeanne Got, Méziane Aite, Ludovic Delage, Jonas Collén, Clémence Frioux, Catherine Leblanc, Simon M. Dittami, Samuel Blanquart, Gabriel V. Markov and Anne Siegel
Genome Research, juin 2023. DOI : https://doi.org/10.1101/gr.277056.122
Contacts
Gabriel Markov, CNRS
gabriel.markov@sb-roscoff.fr
Twitter : @gabrielvmarkov
Laboratoire de Biologie Intégrative des Modèles Marins (CNRS / Sorbonne Université),
Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff
Anne Siegel, CNRS
anne.siegel@irisa.fr
Twitter : @DylissTeam
Institut de recherche en informatique et systèmes aléatoires (CNRS /Université de Rennes).
IRISA, Campus universitaire de Beaulieu - 263 Avenue du Général Leclerc - CS 74205, 35042 RENNES Cedex - France